Доброкачественная наследственная хорея (также известна как доброкачественная семейная хорея) является редким аутосомно-доминантным заболеванием, характеризующимся дебютом в детском возрасте и незначительным прогрессированием двигательных симптомов, в большинстве случаев не сопровождающееся когнитивным дефицитом и психическими расстройствами. [4]
Историческая справка
- Впервые расстройство было описано в 60-х годах, однако, ввиду отсутствия специфических тестов для подтверждения диагноза существование заболевания как самостоятельной нозологической единицы было поставлено под сомнение.
- В 1967 году было обследовано несколько голландских семей, в которых хореиформный гиперкинез отмечался с детского возраста, также членам этих семей было проведено нейропсихологическое обследование, в результате которых не было выявлено отклонений в психической развитии и когнитивных функциях. При составлении родословной был установлен аутосомно-доминантный тип наследования. В тот же период было предположено, что расстройство связано с 14-й хромосомой.
- В 2002 году была изучена итальянская семья с аналогичными клиническими данными и также сделан вывод об участии 14-й хромосомы. После детального изучения хромосомы генетики выявили делецию ДНК в гене NKX2-1, которая приводит к повреждению и утрате белка TITF-1.
- Таким образом, был доказан семейный характер заболевания и установлена его генетическая природа. [8]
- В в 2005 г. M. A. Willemsen и соавт. Был предложен термин "синдром "мозг - легкие - щитовидная железа" с описанием клинического случая о 23-летнем пациенте с легочным альвеолярным протеинозом, первичным гипотиреозом, задержкой моторного развития, выраженной мышечной гипотонией и сформировавшейся еще в детстве хореей. При генетическом обследовании у данного пациента была выявлена инсерционная мутация гена NKX2-1, приводящая к сдвигу рамки считывания белка TTF-1. [11]
Генетические аспекты
- Аутосомно-доминантный тип наследования означает, что:
- мужчины и женщины с одинаковой вероятностью могут быть носителями мутации
- заболевание передается как по мужской, так и по женской линии
- в случае гетерозиготного носительства мутации, заболевание будет унаследовано 50 % потомства; в случае гомозиготного носительства — заболевание наследуется всем потомством.
- Ген NKX2-1 кодирует белок TITF-1, первоначально идентифицированный как тиреоид-специфический фактор транскрипции, регулирующий экспрессию тиреоид-специфических геной, а также участвующий в морфогенезе. Клинически доказано его участие в развитии доброкачественной наследственной хореи, врождённом гипотиреозе и респираторным дистресс-синдроме новорожденных. [1]
- Только в 13% случаев мутация приводит к изолированному расстройству движений, как правило, заболевание, помимо неврологических расстройств, включает субклинические или легкие проявления патологии щитовидной железы и легких. [3]
- Ген NKX2-1 экспрессируется во время эмибрионального развития в щитовидной железе, промежуточной мозге, пневмоцитах II типа, клетках Клара дыхательной системы, поэтому существует более тяжелый аллельный вариант заболевания, сочетающий хореиформный гиперкинез, гипотиреоидизм и неонататальную дыхательную недостаточность.
Эпидемиология
- Распространённость доброкачественной наследственной хореи – как минимум, 0,1- 1: 500 000
- С учётом дефицита диагностики данной патологии показатель распространённости занижен
Классификация доброкачественной наследственной хореи
- На сегодняшний день единой классификации не существует
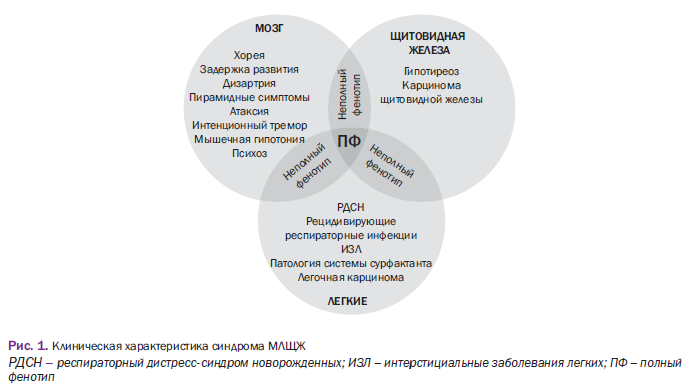
- По клиническим фенотипам можно выделить следующие варианты заболеваний в зависимости от «полноты» синдрома «мозг-лёгкие-щитовидная железа»:
- Полный фенотип, сочетающий
- неврологические симптомы (хорея, дизартрия, задержка развития, пирамидные симптомы, атаксию, интенционный тремор, мышечную гипотонию),
- патологию щитовидной железы ( гипотиреоз, карцинома ЩЖ),
- заболевания лёгких (рецидивирующие респираторные инфекции, интерстициальные заболевания легких, патологию системы сурфактанта, легочную карциному).
- Неполные фенотипы, включающие только 1 или 2 симптомокомплекса со стороны нервной системы, ЩЖ или легких [2]
Клиническая картина
Диагностика
Дифференциальная диагностика
Лечение
Литература
Ограниченный доступ
Для просмотра статьи необходимо
зарегистрироваться
или
авторизоваться